1000 Genomes Project
On this page
History
HapMap Project

Source: Broad Institute
The International HapMap Project was a collaborative effort launched in 2002 to create a haplotype map of the human genome. The project aimed to identify and catalog common patterns of genetic variation (haplotypes) across different populations to facilitate association studies.
- HapMap Phase 1 (2005): Characterized ~1 million SNPs across 269 individuals from 4 populations (YRI, CEU, CHB, JPT)
- HapMap Phase 2 (2007): Expanded to ~3.1 million SNPs across the same populations
- HapMap Phase 3 (2010): Significantly expanded to include ~1.6 million SNPs across 1,184 individuals from 11 populations, providing better global representation
The HapMap project was instrumental in: - Identifying tag SNPs for genome-wide association studies - Understanding patterns of linkage disequilibrium across populations - Providing reference data for imputation methods - Establishing the foundation for population genetics research
1000 Genomes Project
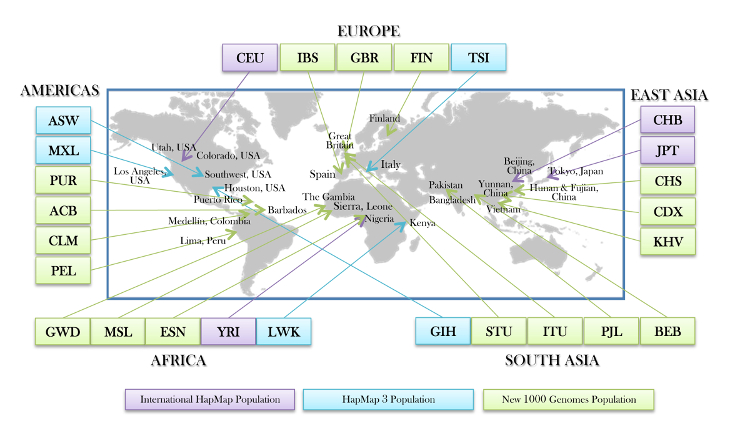
Source: 1000 Genomes Project / IGSR
The 1000 Genomes Project was launched in 2008 as a follow-up to HapMap, with the goal of creating a comprehensive catalog of human genetic variation at higher resolution. Unlike HapMap, which focused on common variants, the 1000 Genomes Project aimed to identify both common and rare variants across diverse populations.
Key milestones:
- Phase 1 (2010): Pilot phase with low-coverage sequencing of 179 individuals from 3 populations
- Phase 2 (2012): Expanded to 1,092 individuals from 14 populations
- Phase 3 (2015): Final phase with 2,504 individuals from 26 populations, providing the most comprehensive catalog of human genetic variation at the time
The 1000 Genomes Project made significant contributions: - Identified millions of previously unknown variants, including rare variants - Provided high-quality reference panels for imputation - Enabled population-specific variant frequency estimation - Facilitated the transition from SNP arrays to sequencing-based studies - Created phased haplotype data for better imputation accuracy
Both projects have been fundamental resources for GWAS, providing reference data that enables researchers to: - Impute untyped variants in their studies - Understand population-specific allele frequencies - Perform fine-mapping and functional annotation - Study population structure and genetic diversity
Why Use 1000 Genomes Project?
The 1000 Genomes Project (1KG) remains one of the most widely used reference panels in genetic research for several key reasons:
1. Comprehensive Variant Catalog
- Contains over 88 million genetic variants (84.7M SNPs, 3.6M indels, 60K structural variants)
- Captures both common (≥1% frequency) and rare variants (0.1-0.5% in coding regions)
- Provides the most complete catalog of human genetic variation across diverse populations
2. High-Quality Imputation Reference
- Phased haplotype data enables accurate imputation of untyped variants in GWAS studies
- Significantly improves statistical power by allowing analysis of variants not directly genotyped
- Widely supported by imputation software (e.g., IMPUTE2, Minimac, Beagle)
- Better imputation accuracy compared to earlier HapMap panels due to larger sample size and sequencing-based data
3. Population Diversity
- Includes 26 populations from 5 major continental groups (Africa, Americas, East Asia, Europe, South Asia)
- Enables population-specific allele frequency estimation
- Supports studies of population structure and admixture
- Allows for population-stratified analyses and ancestry-specific variant discovery
4. Publicly Available and Well-Documented
- Free and open access - no restrictions on data usage
- Available through multiple platforms (IGSR, EBI, NCBI, AWS)
- Comprehensive documentation and metadata
- Active community support and regular updates
5. Integration with Other Resources
- Compatible with major databases (dbSNP, ClinVar, gnomAD)
- Used as reference in many population genetics databases
- Integrates well with functional genomics datasets (ENCODE, Roadmap Epigenomics)
Common Use Cases:
- GWAS imputation: Imputing untyped variants in case-control or quantitative trait studies
- Population genetics: Studying genetic diversity, population structure, and demographic history
- Variant annotation: Determining population-specific allele frequencies for variant interpretation
- Fine-mapping: Identifying causal variants in association regions
- Quality control: Using population frequencies to filter variants and assess data quality
- Ancestry analysis: Estimating genetic ancestry and population admixture
Key terms
HapMap, 1000 Genomes Project (1KG), Ancestry
Download data
HapMap Phase 3
- NCBI FTP (all HapMap releases): https://ftp.ncbi.nlm.nih.gov/hapmap/
- Phase III genotypes (2010-05 release): https://ftp.ncbi.nlm.nih.gov/hapmap/genotypes/2010-05_phaseIII/
- Overview and documentation: NCBI dbGaP / HapMap
1000 Genomes Project (1KG)
- IGSR — browse samples and data: https://www.internationalgenome.org/data
- EBI FTP (project files, VCFs, panels): https://ftp.1000genomes.ebi.ac.uk/vol1/ftp/
- NCBI FTP mirror: https://ftp-trace.ncbi.nih.gov/1000genomes/ftp/
References
HapMap Project
-
International HapMap Consortium. A haplotype map of the human genome. Nature 437, 1299-1320 (2005). doi:10.1038/nature04226
-
International HapMap Consortium. A second generation human haplotype map of over 3.1 million SNPs. Nature 449, 851-861 (2007). doi:10.1038/nature06258
-
International HapMap 3 Consortium. Integrating common and rare genetic variation in diverse human populations. Nature 467, 52-58 (2010). doi:10.1038/nature09298
1000 Genomes Project
-
1000 Genomes Project Consortium. A map of human genome variation from population-scale sequencing. Nature 467, 1061-1073 (2010). doi:10.1038/nature09534
-
1000 Genomes Project Consortium. An integrated map of genetic variation from 1,092 human genomes. Nature 491, 56-65 (2012). doi:10.1038/nature11632
-
1000 Genomes Project Consortium. A global reference for human genetic variation. Nature 526, 68-74 (2015). doi:10.1038/nature15393