Winner's curse
On this page
Introduction
Winner's curse refers to the phenomenon that genetic effects are systematically overestimated by thresholding or selection process in genetic association studies.
When we select variants based on significance thresholds (e.g., \(p < 5 \times 10^{-8}\)), the observed effect sizes for these selected variants tend to be inflated compared to their true effect sizes. This bias occurs because we're conditioning on the variants that passed the significance threshold, which are more likely to have observed effects that are larger than their true effects due to random sampling variation.
Winner's curse in auctions
This term was initially used to describe a phenomenon that occurs in auctions. The winning bid is very likely to overestimate the intrinsic value of an item even if all the bids are unbiased (the auctioned item is of equal value to all bidders). The thresholding process in GWAS resembles auctions, where the lead variants are the winning bids.
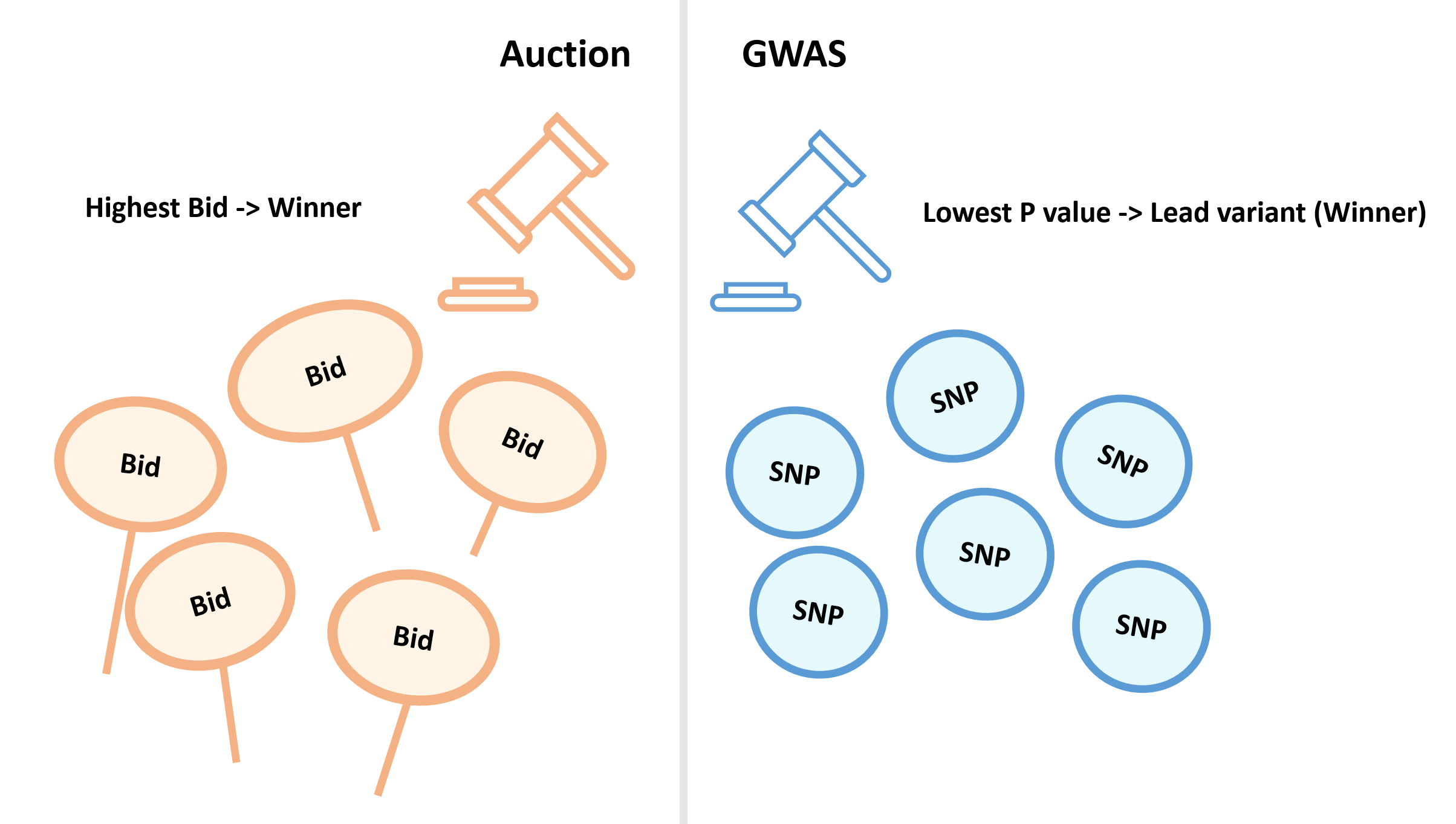
Reference:
- Bazerman, M. H., & Samuelson, W. F. (1983). I won the auction but don't want the prize. Journal of conflict resolution, 27(4), 618-634.
- Göring, H. H., Terwilliger, J. D., & Blangero, J. (2001). Large upward bias in estimation of locus-specific effects from genomewide scans. The American Journal of Human Genetics, 69(6), 1357-1369.
Why does winner's curse matter?
- Effect size estimation: Inflated effect sizes can mislead downstream analyses such as polygenic risk scores (PRS) or Mendelian randomization
- Replication studies: Overestimated effects can lead to failed replications in independent cohorts
- Biological interpretation: Accurate effect sizes are crucial for understanding the true magnitude of genetic associations
- Power calculations: Biased effect estimates can affect power calculations for future studies
Mathematical framework
Asymptotic distribution (unconditional)
The unconditional asymptotic distribution of \(\beta_{Observed}\) (before selection) is:
An example of distribution of \(\beta_{Observed}\)
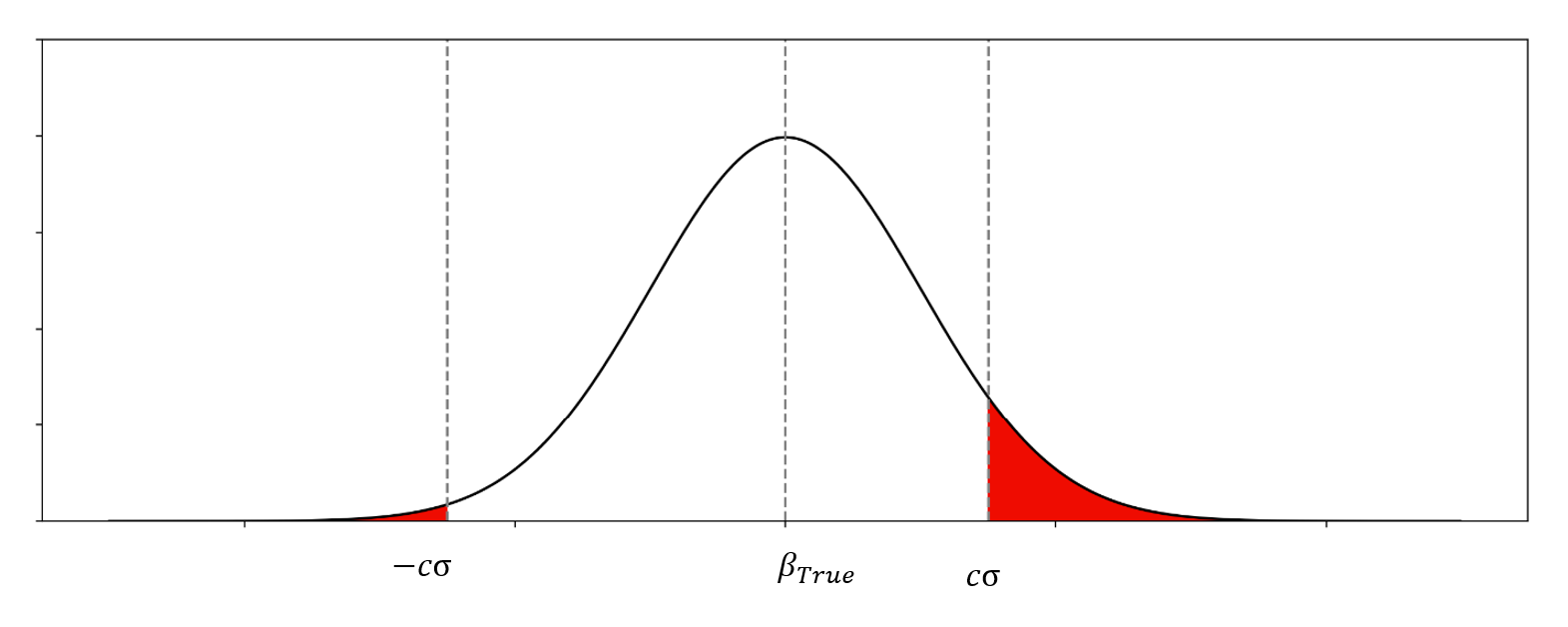
where: - \(\beta_{True}\): The true (unbiased) effect size - \(\sigma\): The standard error of the effect estimate - \(c\): Z score cutpoint corresponding to the significance threshold (e.g., \(c = 5.45\) for \(p = 5 \times 10^{-8}\))
It is equivalent to:
An example of distribution of \({{\beta_{Observed} - \beta_{True}}\over{\sigma}}\)
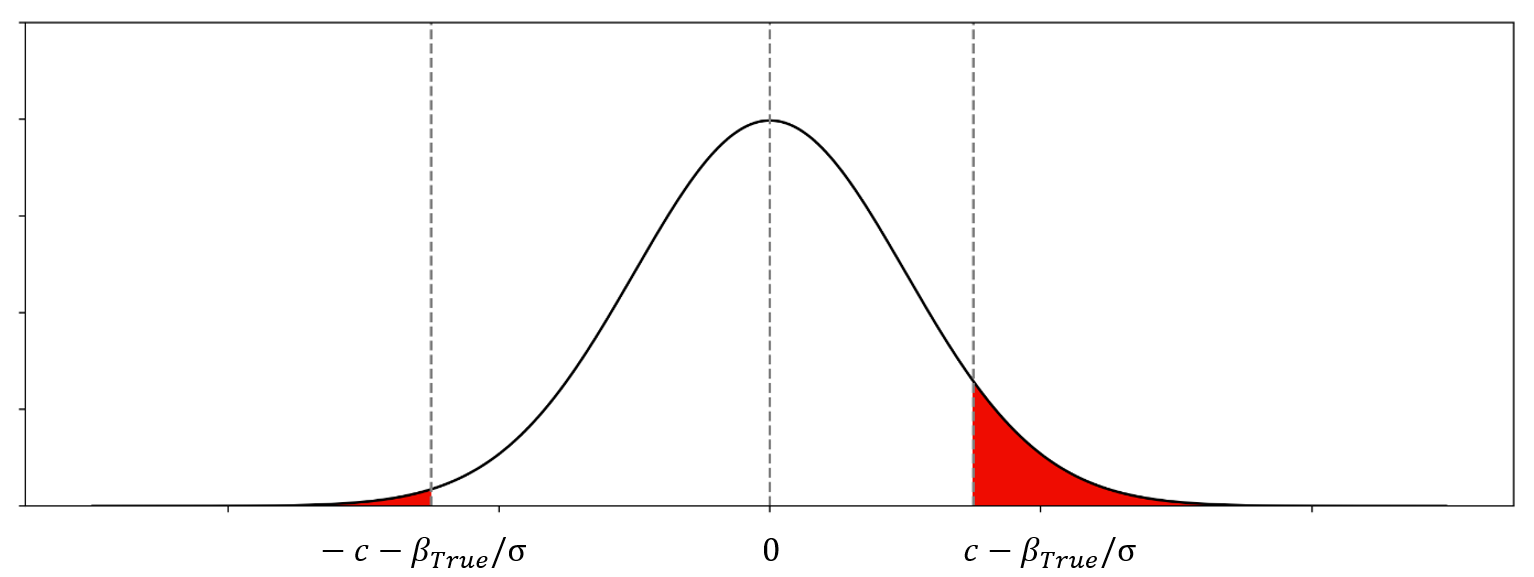
Selection-conditional distribution (two-sided truncated normal)
When we condition on selection (i.e., variants that passed the significance threshold), the distribution of \(\beta_{Observed}\) becomes a two-sided truncated normal distribution (also called a selection-conditional normal distribution). This is a mixture of two tails: the upper tail for positive effects and the lower tail for negative effects.
The conditional distribution of \(\beta_{Observed}\) given selection (i.e., \(|\beta_{Observed}/\sigma| \geq c\)) is:
when
where: - \(\phi(x)\): standard normal density function - \(\Phi(x)\): standard normal cumulative density function - The denominator \(\Phi({{{\beta_{True}}\over{\sigma}}-c}) + \Phi({{{-\beta_{True}}\over{\sigma}}-c})\) represents the probability of selection (the sum of probabilities in both tails)
Expected bias
From the selection-conditional distribution, the expectation of effect sizes for the selected variants (i.e., \(E[\beta_{Observed} | \text{selected}, \beta_{True}]\)) can then be approximated by:
Key observations: - \(\beta_{Observed}\) is biased upward (for positive effects) or downward (for negative effects) - The bias is dependent on \(\beta_{True}\), SE \(\sigma\), and the selection threshold \(c\) - The bias is larger when the standard error is larger relative to the true effect size
Derivation of this equation
The derivation of this equation can be found in the Appendix A of Ghosh, A., Zou, F., & Wright, F. A. (2008). Estimating odds ratios in genome scans: an approximate conditional likelihood approach. The American Journal of Human Genetics, 82(5), 1064-1074.
Winner's curse correction
Important: Only apply to selected variants
This correction is intended only for selected hits (those satisfying the significance threshold).
The correction uses the selection-conditional distribution (two-sided truncated normal), which only applies when variants have been selected based on passing a significance threshold. Without selection, there is no conditioning/truncation, and the unconditional distribution \(N(\beta_{True}, \sigma^2)\) applies. Applying the correction to non-selected variants would be mathematically incorrect and could introduce bias.
To correct for winner's curse, we need to solve for \(\beta_{True}\) given the observed \(\beta_{Observed}\) and standard error \(\sigma\), conditional on the variant being selected. This is done by finding the value of \(\beta_{True}\) that satisfies:
where the expectation is taken over the selection-conditional distribution. This requires solving a nonlinear equation, typically using numerical methods.
Implementation in Python
Winner's curse correction function in Python
import scipy.stats as sp
import scipy.optimize
import numpy as np
def wc_correct(beta, se, sig_level=5e-8):
"""
Correct for winner's curse bias in effect size estimates.
Parameters:
-----------
beta : float or array-like
Observed effect size(s)
se : float or array-like
Standard error(s) of the effect size(s)
sig_level : float
Significance threshold (default: 5e-8 for genome-wide significance)
Returns:
--------
float or array
Corrected effect size(s)
"""
# Calculate z-score cutpoint
c2 = sp.chi2.ppf(1 - sig_level, df=1)
c = np.sqrt(c2)
def bias(beta_T, beta_O, se):
"""Calculate the bias for a given true effect size."""
z = beta_T / se
numerator = sp.norm.pdf(z - c) - sp.norm.pdf(-z - c)
denominator = sp.norm.cdf(z - c) + sp.norm.cdf(-z - c)
return beta_T + se * numerator / denominator - beta_O
# Handle both scalar and array inputs
if np.isscalar(beta):
beta_corrected = scipy.optimize.brentq(
lambda x: bias(x, beta, se),
a=-100, b=100,
maxiter=1000
)
else:
beta_corrected = np.array([
scipy.optimize.brentq(
lambda x: bias(x, b, s),
a=-100, b=100,
maxiter=1000
)
for b, s in zip(beta, se)
])
return beta_corrected
# Example usage
beta_obs = 0.15
se = 0.02
beta_corrected = wc_correct(beta_obs, se)
print(f"Observed effect: {beta_obs:.4f}")
print(f"Corrected effect: {beta_corrected:.4f}")
print(f"Bias: {beta_obs - beta_corrected:.4f}")
Implementation in R
Winner's curse correction function in R
WC_correction <- function(BETA, # Effect size (vector)
SE, # Standard Error (vector)
alpha=5e-8){ # Significance threshold
# Calculate z-score cutpoint
Q <- qchisq(alpha, df=1, lower.tail=FALSE)
c <- sqrt(Q)
# Bias function
bias <- function(betaTrue, betaObs, se){
z <- betaTrue / se
num <- dnorm(z - c) - dnorm(-z - c)
den <- pnorm(z - c) + pnorm(-z - c)
return(betaObs - betaTrue - se * num / den)
}
# Solve for true beta
solveBetaTrue <- function(betaObs, se){
result <- uniroot(
f = function(b) bias(b, betaObs, se),
lower = -100,
upper = 100
)
return(result$root)
}
# Apply correction to all variants
BETA_corrected <- sapply(
1:length(BETA),
function(i) solveBetaTrue(BETA[i], SE[i])
)
return(BETA_corrected)
}
# Example usage
beta_obs <- 0.15
se <- 0.02
beta_corrected <- WC_correction(beta_obs, se)
cat("Observed effect:", beta_obs, "\n")
cat("Corrected effect:", beta_corrected, "\n")
cat("Bias:", beta_obs - beta_corrected, "\n")
Applying correction to GWAS summary statistics
Correcting multiple variants from GWAS summary statistics
import pandas as pd
# Load GWAS summary statistics
sumstats = pd.read_csv("gwas_results.tsv", sep="\t")
# IMPORTANT: Filter for significant variants BEFORE applying correction
# The correction uses the selection-conditional distribution, which only
# applies to variants that passed the significance threshold
significant = sumstats[sumstats["P"] < 5e-8].copy()
# Apply winner's curse correction (only to selected variants)
significant["BETA_corrected"] = wc_correct(
significant["BETA"].values,
significant["SE"].values,
sig_level=5e-8
)
# Calculate the bias
significant["Bias"] = significant["BETA"] - significant["BETA_corrected"]
# Display results
print(significant[["SNP", "BETA", "BETA_corrected", "Bias", "SE", "P"]].head())
When to apply winner's curse correction
- Before replication studies: Correct effect sizes to get more accurate estimates for power calculations
- Before PRS construction: Use corrected effect sizes for more accurate polygenic risk scores
- Before Mendelian randomization: Corrected effect sizes improve MR estimates
- For meta-analysis: Correct effect sizes from discovery studies before meta-analysis
Available tools and packages
Several tools and packages are available for winner's curse correction:
R packages
- winnerscurse: R package providing multiple methods for winner's curse correction
- Installation:
install.packages("winnerscurse") - Documentation: https://amandaforde.github.io/winnerscurse/
- Methods include: conditional likelihood, FDR inverse quantile transformation, and bootstrap-based methods
Python packages
- Custom implementation (as shown above) using
scipy - The
winnerscurseR package can also be used viarpy2in Python
Comparison of methods
Different methods for winner's curse correction have been developed: - Conditional likelihood approach: Used in the examples above (Zhong & Prentice, 2008; Ghosh et al., 2008) - FDR inverse quantile transformation: Alternative approach that may be more robust - Bootstrap methods: Can be computationally intensive but may handle complex scenarios better
For a comprehensive comparison, see: https://amandaforde.github.io/winnerscurse/articles/winners_curse_methods.html
Limitations and considerations
Important considerations
- Assumptions: The correction assumes that the asymptotic normal distribution holds and that variants are independent
- LD structure: The correction may not fully account for linkage disequilibrium (LD) between variants
- Multiple testing: The correction is typically applied per-variant; accounting for multiple testing may require additional considerations
- Effect direction: The correction works for both positive and negative effects, but the bias direction differs
- Small effects: For very small true effects, the correction may be less reliable
Best practices
- Apply correction only to variants that passed the significance threshold: The correction uses the selection-conditional distribution, which only applies to selected variants. Without selection, there is no truncation/conditioning, so the correction should not be applied.
- Use the same significance threshold for correction as was used for selection
- Consider the standard error when interpreting corrected effects
- Validate corrected effects in independent replication cohorts when possible
References
Methodological papers
-
Zhong, H., & Prentice, R. L. (2008). Bias-reduced estimators and confidence intervals for odds ratios in genome-wide association studies. Biostatistics, 9(4), 621-634. https://doi.org/10.1093/biostatistics/kxn001
-
Ghosh, A., Zou, F., & Wright, F. A. (2008). Estimating odds ratios in genome scans: an approximate conditional likelihood approach. The American Journal of Human Genetics, 82(5), 1064-1074. https://doi.org/10.1016/j.ajhg.2008.03.002
Review and comparison papers
- Forde, A., & Wade, K. H. (2022). Winner's Curse Correction and Variable Thresholding Improve Performance of Polygenic Risk Modeling Based on Genome-Wide Association Study Summary-Level Data. PLoS Genetics, 18(6), e1010173. https://doi.org/10.1371/journal.pgen.1010173
Software and resources
- winnerscurse R package: https://amandaforde.github.io/winnerscurse/
- Comparison of winner's curse methods: https://amandaforde.github.io/winnerscurse/articles/winners_curse_methods.html